Protecting Pharmaceuticals at the Intersection of Patent and Regulatory Law
Mar 14th, 2018 by John Wizeman | News | Recent News & Articles |
 Over three decades ago, the United States Congress passed the Drug Price Competition and Patent Term Restoration Act[1]. This piece of legislation, known as the Hatch-Waxman Act, tackled the difficult task of protecting pharmaceutical innovator intellectual property while ultimately providing increased competition and decreased cost to consumers through accessible generic drugs. This legislative task was accomplished with two pieces of intersecting laws: (i) the patent provisions under 35 USC which provide for up to five additional years of patent term extension and (ii) the drug exclusivity provisions under 21 USC 355 which provide certain regulatory and marketing exclusivity periods upon drug approval. The intersection of these patent and regulatory/marketing exclusivity periods provide innovator drug developers with a net exclusivity period. Given the immense monetary and time investment for developing new drugs, maximizing this net exclusivity should be a major focus of patent practitioners in the pharmaceutical field. By maximizing this net exclusivity, innovator drug developers can recoup their investment, as well as provide a stable foundation and incentive for continued drug discovery. To understand how to maximize this window, those involved need to understand the important role this intersection of patent and regulatory law holds.
Over three decades ago, the United States Congress passed the Drug Price Competition and Patent Term Restoration Act[1]. This piece of legislation, known as the Hatch-Waxman Act, tackled the difficult task of protecting pharmaceutical innovator intellectual property while ultimately providing increased competition and decreased cost to consumers through accessible generic drugs. This legislative task was accomplished with two pieces of intersecting laws: (i) the patent provisions under 35 USC which provide for up to five additional years of patent term extension and (ii) the drug exclusivity provisions under 21 USC 355 which provide certain regulatory and marketing exclusivity periods upon drug approval. The intersection of these patent and regulatory/marketing exclusivity periods provide innovator drug developers with a net exclusivity period. Given the immense monetary and time investment for developing new drugs, maximizing this net exclusivity should be a major focus of patent practitioners in the pharmaceutical field. By maximizing this net exclusivity, innovator drug developers can recoup their investment, as well as provide a stable foundation and incentive for continued drug discovery. To understand how to maximize this window, those involved need to understand the important role this intersection of patent and regulatory law holds.
Why is Exclusivity Important?
New drug development takes a long time, as products can be in clinical trial and under regulatory review during much of the duration of the term of the originally filed patents covering the drug. In other words, the effective patent term is ticking away, and time is money. The average cost of drug research and development is estimated to be between ~$650 million[2] and $2.5 billion[3], or even higher[4]. In addition to research costs, the estimated proportion of drugs that make it through clinical trials hovers down near one in ten[5,6]. While consumer cost is a major consideration for allowing generic drugs to market, innovator drug companies often need time to recoup their monetary investment. However, the revenue-producing period is drastically diminished after patent expiry [7]. Hatch-Waxman provides several regulatory and marketing exclusivity periods after the approval of the innovator drugs, for example new chemical entities (NCEs), as well as provisions relating to their generic equivalents. Hatch-Waxman also provides the regulatory framework for innovators to recover a portion of the patent term lost to clinical development and regulatory review.
How can Net Exclusivity be Maximized?
Exclusivity for new drug products can be extended beyond the original patent term through both patent term extension[8] and through regulatory and marketing exclusivity [9]. Patent term extension (or more accurately, “restoration”) can provide a means to recover part of the patent term lost during drug development and FDA review. Regulatory/marketing exclusivity, which seemingly operates independently from patent exclusivity, can overlap with or add exclusivity after the end of the patent term. Key to navigating the intersection between patent and regulatory law and to maximizing exclusivity is to maximize each individual exclusivity period with minimal overlap. For instance, patent term restoration must be applied for prior to patent expiry, but both the original patent term and any extension can overlap with the regulatory/marketing exclusivity periods. This results in a loss of actual protection, or the effective patent term, which in turn cuts into the important revenue-producing lifespan of the pharmaceutical product. Complicating this factor is the unpredictable nature of clinical trials, which can extend for many years while the patent term runs out.
What Factors are Considered for Patent Term Restoration?
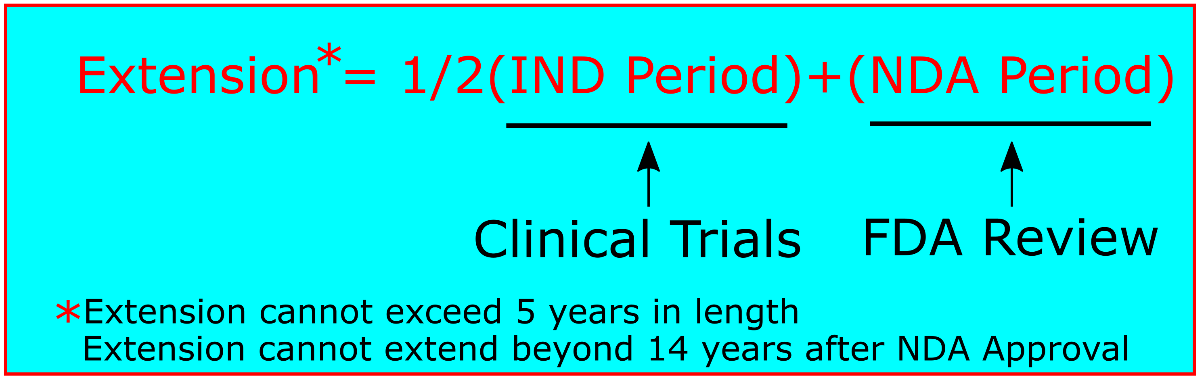
While the actual time period in which a drug product undergoes clinical trials may vary, a simple calculation is used to determine the length a patent term may be extended. As long as the applicant has diligently pursued the clinical development and regulatory approval, patent term restoration allows for the equivalent of one half of the clinical trial period (after the IND is filed, but before the NDA is filed) in addition to the entire review period after clinical trials have concluded (after the NDA is filed, but before approval). This calculation can be expressed as Extension = ½(IND)+(NDA).[10]. Two limitations, however, apply to this calculation. First, the original patent term with the extension cannot extend beyond 14 years after FDA approval of the NDA. Second, the extension term itself cannot be more than 5 years.
What About Regulatory/Market Exclusivity?
The other consideration for the extension of exclusivity is the type of product and what regulatory and marketing exclusivity is applicable. For NCEs, regulatory exclusion provides for a 5 year period during which no Abbreviated New Drug Application (ANDA, an application for a generic drug) can be submitted. In certain cases, this may be shortened to 4 years, but the additional 1 year of marketing exclusivity still remains. Drug products based on NCEs that have already been approved may warrant further regulatory review and protection, such as new dosages, new dosage forms, and new indications. In this case, innovators may file a supplemental NDA. Approved supplemental NDAs provide innovators with a 3 year exclusivity window[11]. In order to encourage clinical trials for pediatric diseases, instead of relying on studies in adults, there is protection for innovators who develop pediatric drugs that undertake appropriate clinical trials. Six months of exclusivity is added to the protection of all “formulations, dosage forms, and indications” [12] for any molecule that underwent pediatric studies. Additional protection is provided for those investing time and resources developing orphan drugs. These are drugs that are targeted to diseases affecting less than 200,000 people in the U.S. If a drug is targeted toward one of these rare diseases, or toward diseases “that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug”, they may be protected for 7 years instead of 5 [13]. Lastly, there is protection for biological products. Biological products, or “biologics”, are mixtures of molecules, often with unknown or incomplete structure and characterization, that are derived from a biological system instead of chemically synthesized like most drugs[14]. Biologics have a protected regulatory period that is effectively 12 years.
Maximizing Exclusivity
Considering the complicated pathway that drug products must be guided along to maximize revenue-producing periods, the goal of those responsible should be to maximize the exclusivity window. While the considerations discussed place emphasis on several clear factors, these become even further complicated when considering products with multiple patents or patents dedicated to multiple products. Maximizing exclusivity relies on the ability to navigate these factors, and should be the focus for patent practitioners, researchers, and innovators.
-John Wizeman, PhD and Anthony Sabatelli, PhD, JD
John Wizeman recently received his Ph.D. in biomedical sciences with a concentration in neuroscience from the University of Connecticut Health Center. His research focused on retinal injury and repair in addition to several student leadership roles. He then moved into a postdoctoral position researching cerebellar development. He has published numerous papers in different specialties of neuroscience. Currently, he focuses in neurodevelopment and teaches several Medical/Dental and Graduate courses at UConn Health.
This article is for informational purposes, is not intended to constitute legal advice, and may be considered advertising under applicable state laws. The opinions expressed in this article are those of the author only and are not necessarily shared by Dilworth IP, its other attorneys, agents, or staff, or its clients.
[2] Prasad V, Mailankody S. (2017) Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Intern Med. 177(11):1569-1575. PubMed PMID: 28892524;
[3] Chakravarthy R, Cotter K, DiMasi JA, Milne C-P, Wendel N. Public and private sector contributions to the research and development of the most transformational drugs of the last 25 years. Boston: Tufts Center for the Study of Drug Development, January 2015 (http://csdd.tufts.edu/files/uploads/PubPrivPaper2015.pdf).
[4] https://www.forbes.com/sites/matthewherper/2013/08/11/how-the-staggering-cost-of-inventing-new-drugs-is-shaping-the-future-of-medicine/#65efc62013c3
[5] http://www.amplion.com/clinical-development-success-rates
[6] Wong CH, Siah KW, Lo AW. (2018) Estimation of clinical trial success rates and related parameters. Biostatistics. PubMed PMID: 29394327.
[7] https://www.forbes.com/sites/johnlamattina/2014/05/01/drug-prices-defy-gravity-until-the-patent-expires/#2684ccfd2639
[8] 35 USC § 156
[9] 21 USC § 355
[10]http://www.fda.gov/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/ucm069959.htm
[11] 21 C.F.R. 314.108
[12] https://www.fda.gov/Drugs/ResourcesForYou/Consumers/ucm143565.htm
[13] https://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
[14] https://www.fda.gov/ForConsumers/ConsumerUpdates/ucm048341.htm